R 讀寫 FASTA 檔
先前曾介紹 R 讀取 FASTQ 檔的方式,本文則要分別介紹如何在 R 環境,使用 seqnir 套件或 baseR 讀取 FASTA 檔。
FASTA 檔
FASTA 格式用於紀錄核酸或胜肽的序列。以基於增幅子的資料處理為例,諸如 unique reads、OTU/ASV list、SILVA/RDP reference sequence 皆是以 FASTA 格式儲存。
在 FASTA 格式 ,一條序列的紀錄由兩部分組成:
- 以 “>” 起頭的描述列,用以區隔不同序列的紀錄,並保存序列的名稱、辨識碼或來源等資訊。
- 由鹼基或胺基酸字符組成的序列字串(鹼基和胺基酸字符的 IUPAC 命名可參考維基百科的簡介)。
典型的 FASTA 格式如下,所屬文件通常會以 .fa、.fasta 或 .fna 為副檔名,但實際並無統一規範。
1 | >OTU1 |
與 FASTQ 檔不同,FASTA 檔的序列字串未必在同一列。依慣例,序列字串長度不會超過 80 個字元(這可能跟方便在終端介面閱讀有關),所以較長的序列會被換行符分隔成好幾列。
使用 Seqinr 處理 FASTA
Seqinr 是專門用於處理核酸和蛋白質序列的套件,其功能枚舉如下:
- 自Genbank等資料庫下載序列
- 讀寫 FASTA 檔
- 計算各式鹼基統計量
- 序列比對
- 轉錄或轉譯等字符抽換
由於 seqinr 存放在 CRAN 庫,所以可以直接輸入install.packages("seqinr")安裝。
1 | # install and read seqinr package |
先來看看我們的示範資料 dna.fasta。為了凸顯 FASTA 檔的序列字串不一定在同一行的特質,我故意將示範資料段設定得這麼混亂。然而,正常的資料雖然列數可能隨序列長度而異,但每一列的字串長度是一致的。
1 | >seq1 |
讀取 fasta 檔
以 seqinr 的 read.fasta 能把 FASTA 檔讀為由字串向量構成的 list。
1 | > dna <- read.fasta("dna.fasta") |
序列簡易操作
統計序列長度分布
1 | # 列出各序列的長度 |
計算 GC content
1 | > sapply(dna, GC) |
將 DNA 序列轉換為胺基酸序列
1 | > translate(dna$seq1) |
繪製序列比對的 dot plot
1 | > dotPlot(dna$seq1, dna$seq3) |
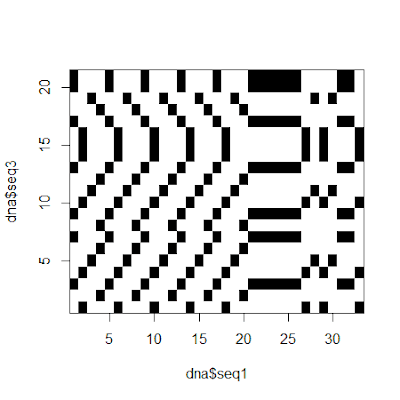
選取特定序列的方法
1 | dna[sapply(dna, GC) > 0.5] \\ 尋找 GC content > 0.5 者 |
儲存 FASTA 檔
1 | write.fasta(dna, names = names(dna), file.out = "dna_rw.fasta", nbchar = 5) |
names:設定序列的名稱,也就是 “>” 後的文字file.out:儲存的檔名與路徑nbchar:序列幾個字符一列
使用 Biostrings 處理 FASTA
除了保存於 CRAN 的 seqinr,存放於 bioconductor 的 Biostrings 也能讀取 FASTA 檔。於 R 終端輸入以下指令即可安裝。
1 | # install and read Biostrings package |
此處,我使用 readDNAStringSet 讀取FASTA檔為 DNAStringSet 物件
1 | > dna <- readDNAStringSet("dna.fasta") |
除了readDNAStringSet,也可以針對序列的類別分別使用 readRNAStringSet 和 readAAStringSet 讀取 FASTA 檔。讀取的方式會影響能套用的 function 數據的存儲與呈現。例如若以 readRNAStringSet 讀取 DNA 序列會自動掠過不符類別的字符。
1 | > readRNAStringSet("dna.fasta") |
另外,亦可使用 readBStringSet 將 FASTA 檔視為一般字串讀取,只是這樣就無法使用某些 function,例如求序列的 reverse complement:
1 | # read .fasta into DNA string set |
選取特定序列則需要調出 DNAStringSet 物件的 range 資料,range 資料是一個 data.frame,紀錄了序列的長度和名稱等資訊。
1 | > dna@ranges |
1 | dna[dna@ranges@width > 25] \\ 保留長度 > 25 bp 的序列 |
而使用 writeXStringSet 儲存 FASTA 檔時,會把示範資料中分為多列的序列貼到同一列。
1 | writeXStringSet(dna, "dna_rw.fasta", format = "fasta") |
Biostrings 其他功能可參考 Biostrings Quick Overview
使用 baseR 處理 FASTA
若不想為了簡單的序列統計和操作而裝套件,也可以使用 baseR 將 FASTA 檔。
讀取 fasta 檔
簡言之,以 readLines 讀取 FASTA 檔所有內容並存為字串向量。接著,使用 grep 搜尋以 “>” 開頭的描述列之索引號,並以 gsub 移除 “>” 取回序列辨識碼。最後利用 paste連接位於描述列的索引號之間的鹼基字串。
1 | > read_fasta <- function (path) { |
序列簡易操作
統計序列長度分布
1 | # 列出各序列的長度 |
繪製序列長度數量分布直方圖
1 | > hist(nchar(dna)) |
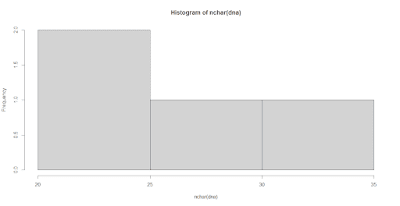
辨識 unique sequence
1 | > unique(dna) |
選取特定序列
1 | dna[nchar(dna) > 25] \\ 保留長度 > 25 bp 的序列 |
儲存 FASTA 檔,即用 names 取出 list 的名稱(也是序列的名稱),再貼上 “>” 依序寫到文件之中。
1 | > write_fasta <- function (obj, path) { |
小結
1 | # 以 seqinr 讀寫 FASTA 檔 |