如何評估基因體結構變異的分析效度?
結構變異(Structural variants, SVs)泛指基因體中長度超過 50 bp 的變異,包含插入 (insertion)、刪除 (deletion)、易位 (translocation) 與倒置 (inversion) 等類型。雖然人類的結構變異總數低於小片段變異,單一變異可能涵蓋影響多個基因片段,影響生理功能甚或引發疾病,使之成為臨床檢測的潛在標的。
隨著定序技術成熟,偵測結構變異的演算法推陳出新。為了能客觀評估這些方法的效度,Genome in a Bottle (GIAB) 以 HG002 樣本,採用多項定序與分析技術,並且透過親屬資料交叉驗證,建立了結構變異的標準資料。
本文將介紹如何使用 Truvari 基於 GIAB 標準資料來評估結構變異的分析效度。除了提供具體的操作方式外,也探討實際分析時可能遇到的技術挑戰。
急用?
假設你們已定序了 HG002 樣本,目前唯一有結構變異(以下簡稱為 SVs)標準資料的樣本,並且使用參考基因體 GRCh37 版分析,獲得 query.sv.vcf.gz 和 query.sv.vcf.gz.tbi。
首先,檢查檔案的格式是否符合 VCF v4.2 規範,每筆紀錄的 INFO 都要如以下範例包含 SVTYPE 和 SVLEN。
1 | #CHROM POS ID REF ALT QUAL FILTER INFO FORMAT NA00001 |
接著從 NCBI 取得 SVs 標準資料 (NIST SVs for HG002),檢查 truth vcf 和 query vcf 的染色體命名慣例(有或沒有帶 “chr” 前綴)。如果兩者不同,需要統一兩個 vcf 的命名方式,作法可參考我之前寫的短文:轉換 VCF 檔染色體命名慣例)。
1 | $ wget https://ftp.ncbi.nlm.nih.gov/ReferenceSamples/giab/data/AshkenazimTrio/analysis/NIST_SVs_Integration_v0.6/HG002_SVs_Tier1_v0.6.vcf.gz |
然後安裝 Truvari,它是 GIAB 團隊用於比較 SVs 的工具。
1 | python3 -m pip install Truvari |
如果你的伺服器有安裝 docker,也能從 biocontainers 下載封裝好的 image。
1 | # https://biocontainers.pro/tools/truvari |
最後指定你的輸出入資料和工作目錄,再透過 docker 執行 truvari。
1 | $ docker run -v $PWD:$PWD -w $PWD quay.io/biocontainers/truvari:5.2.0--pyhdfd78af_0 bench -b HG002_SVs_Tier1_v0.6.vcf.gz -c <query>.sv.vcf.gz -o <outdir> ---includebed HG002_SVs_Tier1_v0.6.bed.gz |
待程式執行完畢(< 15 分鐘),查看輸出目錄的 summary.json 了解結果。完整的結果說明可參考 Truvari 的官方說明,我自己會觀察以下幾個數值:
- base cnt: truth vcf 的變異總數。NIST_SVs 位於可靠區間的變異約有 9000 多筆,如果數據差太多,可能是調整 truth vcf 的格式時出錯,或是用到錯誤的置信區間 BED 檔。
- TP-comp:query vcf 與 truth vcf 相符的變異總數。如果數值為零或很低,可能是因為染色體編號格式不符,或是 query & truth vcf 使用了不同的參考基因體。
- precision, recall, f1:數值越接近 1 表示效度越好。
目前單一分析工具的 F1 介於 50% - 80% 之間,所以數值不理想是正常現象。畢竟,SVs 橫跨的範圍大,序列定位與比對的精準度受限於次世代定序的長度限制。
跑完了,我該注意些什麼嗎?
有,目前評估 SVs 分析的資料與方法有限,因為:
- 目前僅 HG002 有共認的 SVs 標準資料,因為它需要用到親屬的數據交叉驗證。
- HG002 的標準資料沒有基因片段重複 (duplication),所以 CNVs 的分析效度限於基因片段刪除。
- NIST SVs 的標準資料基於 GRCh38。如果你的資料使用其他版本的基因體,需要自行使用 leftalign 調整。
- 雖然臨床應用更在意位於 exon 的變異,標準資料仍包含了位於 intron 的變異。
如果上述限制不符合需求,可以考慮參考 GIAB 的方法自行建立資料集,就像其他想要研究 HG001 SVs 的研究團隊(例如 NA12878_PacBio_MtSinai)。然而,由於成本考量,自建資料通常不如標準資料嚴謹。
結構變異和小片段變異的效度評估有什麼差異?
結構變異和小片段變異的效度評估的方法差異源於尺度不同。同一個變異在 truch & query vcf 可能因為序列定位、比對和表示方法差異而略有不同,如果要求兩者完全相符,可能高估變異分析的錯誤率。由於小片段變異的長度多半在 16 bp 以下,變異表示法的些微誤差都會導致誤判,所以需要一系列的標準化甚至局部重新比對來辨識相同變異的不同形式。
然而,結構變異──你感受看看吧,這是標準資料其中一個長度為 2358 bp 的插入序列:
1 | 1 724861 HG2_Ill_breakscan11_2919 G GAATGGAATGCAATGGAATGCACTCGAACGGATTGGAATGGAATGGACTCGAATAGAATGGAATAAAATGAAATGGACTCCAATGGATTGGAATGGAATTGACTCCAATGGAATTGAATGGAGTGGAACCGAATGGAACGGATTGGAATGGAATGCACTCGAAATGAATTTGAATGGAATGGATTGGGCTCAAATGGAATGGAATGGAATGGAATGGAATGGAATGAACTCAAATGGATTAGCATGGAATGAAGTGGACTCGAATACAATGGAATGGAATGGACTCGAATGGAATGGAACGGACTTGAACGGAATGGAGTGGAATGGACTCGAATGGAATGGAGTTGAATGGACTCGAATGGAATGGAATGTAAAGGAATGGAATGAACTCGAAAGGAGTGGAATGTAATGGAATGAAATGGACTCGAATGGAATTAAATGGAATGGAACGGAATGGACTGGGATGGAATGGAACGGAACGGAACGCAGTTGAATTGAACGGACCCGGAATGGAATGGAATGGAATGGAATGAAATGGAATGAAGTGGACTCTAATGGAATGGAATGGAATGGAATGGAATGGAATGGAATGGAATGGAATGGAATGGAATGGAATAGACTCGAATGAAATGGGATGGACTCGAATGGAATGGAACGGAATGGAATCGACTCGAGTGGAATGGAATGGAATGGAATGGAATGGAATGGAATGGAATGGAATGGAATAGAATGGAATTGACTCAATTTAAATGGAATGAAGTGGAATGAACTCGAATGGCATGCAATGGAATGGAATAGAATCGAATGGAATAGAATGGACCCAAATGGAATGGAACGGAATGGAATGGAATGGAACGGAATGGAATAGAACGGAACGGAATGGAATGGATTGGAATGAACTCCAACGGAATGGAATGGACTCGAATGCAATGGAATGGAATGGAATGGAATGGAGTGGACTGGAATGGAATAGAATGGAATGGAATGGATAGGACTGGAATGAAATGGAATGGAATGGACTCGAATGGAATGGAATGGAATGGAATGGACTCAAATGGAATGGAATGGAATGGAATGGACACGAATGGAATGGAATTGAATGGAATGGAATGGACTGTATGAAAAGGAATGGATTGGAAAGGAATGGAATAGAACGGAATGGACTCGAATGGAATGGAAAGGACTCGAGTGGAATGGAATGGAATGGAATGGACTCGAATGGAATGGAGTGGAATGTATGCGAATGGAATGGAATTGAATGGATTCGAGTCTAACGGAATGTATGGAATGGACTCGAATGGAATGTAATGTAATGGAATGAAATGGACGCGAATGGAATGGAATGGAATGGAATGGAATGGAGTGGAATGGAATGGACTCGAATGGTATGGAATGGAATTGAATGGACTCGATAGGAATGGAATGGAATGGATTGGACTCGAAAGGAATGTAATGGAATGAAATGTGCTGGAATGGAATGGAATGGAATGGAATAAAATGTAATGGAATGGACTCGAATGGAATACAGTTGAATTGAATGGACCCGAAAGCAATGGAATGGAATGGAACGGATTGGAAGGGAATGGAATGGAATGAAATGGAAAAGACTCGAATGGAATGGAATGGACTCGAATGAAATGGAGTGGACTAGAATGGAATGGAATGGACTTGAAAGAAATGGAATGCAGTGGAATGGACTCGAATGGAATGCAATGGAATGGAATAGACTCGAACGGAATGGAATGGAATGGAATGGAATGGAATGGAATGGAATGGAATGGAACGGAATGGACGCGAATGAAATGGAACGGAACGGAATGGACTCGGATGGAACGGAATGGAACGGAATGGAATGGAATTTACTCGAATGGGATGGAATGGAATGGAATTTACTCGAATGGAATGGAATGGAATGGACTGAAATGGAATAGCATGGAATGGAATGGACTCGAATGCAATGGAATGGAATGGACTCGAATGGAACGGAATGGACTCGAACGGAGTGGAGTCGAATGGATTCGAATGGAATGCAATGGAATGGAACGGAATGCAATGTACTCGAATGGAATGGAATGTAATAGAATGAAAATTACTCGAATGGAATGGAATGGAATGGACTCCAATGGAATGGAATCGAACGGACTCGAATAGAATGCAGTTGAATTGAATGGACCTGAAAGAATGCAATGGAATGGAATGAAATGGACTCGAATGGAATGGAATAGACTGAAATGAAATGGAATGTACTGGAATGGAATGGAATGGAATGTACTGGAATGGAATGGAATGGACTCGAATGATATGCAATTGAATGGACTCGCATGGATTGGAATGGACTCTAGTGGAATGGAATGGAATA 20 NoConsensusGT ClusterIDs=HG4_PB_HySA_1:HG4_PB_HySA_2:HG2_Ill_breakscan11_2919:HG2_PB_SVrefine2Falcon1plusDovetail_10:HG2_10X_SVrefine210Xhap12_1:HG4_PB_SVrefine2Falcon1Dovetail_3;NumClusterSVs=6;ExactMatchIDs=HG2_Ill_breakscan11_2919;NumExactMatchSVs=1;ClusterMaxShiftDist=0.0156118143459916;ClusterMaxSizeDiff=0.0151898734177215;ClusterMaxEditDist=0.0256788044622185;PBcalls=4;Illcalls=1;TenXcalls=1;CGcalls=0;PBexactcalls=0;Illexactcalls=1;TenXexactcalls=0;CGexactcalls=0;HG2count=3;HG3count=0;HG4count=3;NumTechs=3;NumTechsExact=1;SVLEN=2358;DistBack=53075;DistForward=7;DistMin=7;DistMinlt1000=TRUE;MultiTech=TRUE;MultiTechExact=FALSE;SVTYPE=INS;END=724861;sizecat=gt1000;DistPASSHG2gt49Minlt1000=.;DistPASSMinlt1000=.;MendelianError=.;HG003_GT=./.;HG004_GT=./.;TRall=TRUE;TRgt100=TRUE;TRgt10k=FALSE;segdup=TRUE;REPTYPE=SIMPLEINS;BREAKSIMLENGTH=1947;REFWIDENED=1:724845-726791 GT:GTcons1:PB_GT:PB_REF:PB_ALT:PBHP_GT:PB_REF_HP1:PB_ALT_HP1:PB_REF_HP2:PB_ALT_HP2:TenX_GT:TenX_REF_HP1:TenX_ALT_HP1:TenX_REF_HP2:TenX_ALT_HP2:ILL250bp_GT:ILL250bp_REF:ILL250bp_ALT:ILLMP_GT:ILLMP_REF:ILLMP_ALT:BNG_LEN_DEL:BNG_LEN_INS:nabsys_svm ./.:./.:./.:3:15:./.:5:36:0:0:./.:0:0:4:18:1/1:1:27:./.:11:120:.:2370:. |
一個結構變異裡面可能混雜定序錯誤及小片段變異,或是嵌套著其他大片段的插入與刪除片段。結構變異的長度增加了評估效度的複雜性,例如要怎麼拆分彼此嵌套的變異?要怎麼評估倒置和易位的變異?相對於涉及結構變異本質的問題,序列比對造成幾個 bp 的差異也顯得微不足道了。換句話說,評估結構變異的分析效度更在意「突變」這件事特徵,而不是少數鹼基的變化程度。
目前,結構變異的分析工具尚未成熟,比較分析結果的手段也仍待開發,學界的共識是限縮分析範圍並且採用模糊比對來迴避問題。首先,因為尚無評估其他變異的公認手段,NIST 的標準資料只納入插入和刪除變異。其次,所有位置彼此重疊的變異被移除了,不用顧慮彼此嵌套的變異。因此,比對 truth & query vcf 的時候能採取較寬鬆的標準以容忍技術雜訊,只要變異的種類相同、位置相鄰、長度相似、序列吻合便能視為 true positive(畢竟鄰近位置沒有其他會造成混淆的變異)。
首先,benchmark 資料裡面只包含插入和刪除兩種變異,降低座標比對的困難度。其次,所有位置彼此重疊的結構性變異都從資料裡移除了,所以分析時就不用考慮複雜性變異的狀況。在限縮了benchmark 資料的規模以後,結構性變異的比較也變得較為簡潔,它仰賴 VCF v4.2 的四項資訊:
CHROM:結構變異所在的染色體編號。POS:結構變異的起始位置。SVTYPE:結構變異的類型。SVLEN:結構變異的長度,插入的符號為+,刪除的符號為-。
只要種類一樣(同屬DEL/INS)、位置相近、長度雷同、序列相似就算是偵測到相同的變異,畢竟 truth 變異周圍可能造成混淆的變異都被移除了。
能否說得具體一點啊?
就拿 Truvari bench 為例吧。Truvari 在比較 truth & query vcf 時,會考慮其中位置相近的數個結構變異,以涵蓋彼此相符的潛在變異。算法的概要如下所示:
- 混合 truth & query vcf,按照染色體編號與基因體位置排序變異。
- 將鄰近的變異劃為同組,以涵蓋可能相符的潛在變異。
- 兩兩比較組內變異,計算相似度指標。
- 如果相似度大於閾值,則視為 true positive;若多組變異皆通過閾值,選擇最相似者。
- 若無符合標準的變異,則 truth 變異視為 false negtive,query 變異視為 false positive。
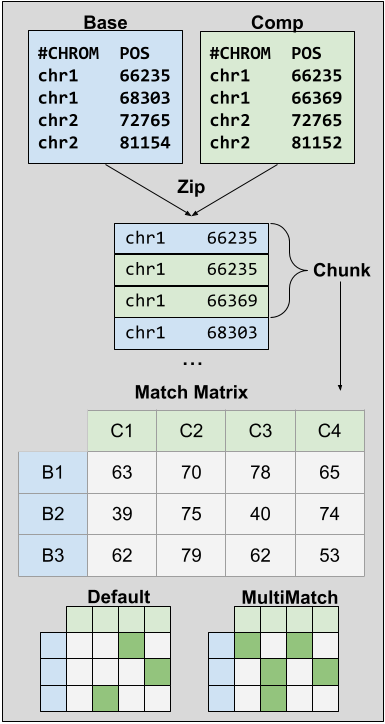
Truvari 評估隸屬同一組的變異時,會先確認 truth & query 是相同變異(插入或刪除),而且彼此的位置有重疊才會計算兩者的相似度。如果認為預設條件太過嚴苛,可以透過 --refdist 選項設定變異兩端的寬限距離。舉例來說,如果要求變異彼此重疊(refdist=0),那麼 truvari 會忽略以下兩個變異。
1 | # 兩者沒有重疊 |
不過,設定 2bp 的寬限距離後( refdist=2),就能保證它們納入計算。
1 | # 另一個變異在寬限範圍內 |
接著,Truvari 會以長度差異、序列相似與重疊比例等特徵評估變異的相似度,
pctseq:插入序列的相似性,公式請參考 English et al. (2022)。pctsize:長度相似性,即「較短的變異長度/較長的變異長度」pctovl:彼此重疊的比例,即「重疊長度/較常的變異長度」
如果想要了解詳細判定過程,執行分析時可加入 --debug 列出每對變異的判定標準與結果。
哪裡可以取得 benchmarking 資料?
GIAB 提供多種結構性變異的標準資料,他們納入 HG002, HG003, HG004 的家族樣本,採用多平台定序與變異分析工具交叉驗證,針對基因體普遍區域 (NIST) 和臨床挑戰區域 (CMRG) 生成可信度較高的標準資料。HG001 則欠缺可供驗證的家族性資料,目前沒有具共識的標準資料。
| Variant | Source | Genome | Version | VCF | BED | Url |
|---|---|---|---|---|---|---|
| Structural variants | NIST | GRCh37 | v0.6 | HG002_SVs_Tier1_v0.6.vcf.gz | HG002_SVs_Tier1_v0.6.bed | NIST_SV_v0.6 |
| Structural variants | CMRG | GRCh37 | v1.00 | HG002_GRCh37_CMRG_SV_v1.00.vcf.gz | HG002_GRCh37_CMRG_SV_v1.00.bed | CMRG_v1.00/GRCh37 |
| Structural variants | CMRG | GRCh38 | v1.00 | HG002_GRCh38_CMRG_SV_v1.00.vcf.gz | HG002_GRCh38_CMRG_SV_v1.00.bed | CMRG_v1.00/GRCh38 |
| Tandem repeats | TandemRepeats | GRCh38 | 1.0 | HG002_GRCh38_TandemRepeats_v1.0.vcf.gz | HG002_GRCh38_TandemRepeats_v1.0.bed.gz | TandemRepeats_v1.0 |
CMRG 的標準資料欠缺 SVTYPE 和 SVLEN,需要自行處理。
1 | chr2 1005806 . AATATATATATATATATATATATATATATATATATATATATATATATATAT A 30 . REPTYPE=SIMPLEDEL;BREAKSIMLENGTH=19;REFWIDENED=chr2:1005807-1005875 GT:AD 0|1:0,1 |
可以用 python 腳本根據 ref 和 alt 的長度來判斷 SVTYPE 和 SVLEN:
SVTYPE:如果ref的長度大於alt就是DEL,反之則是INS。SVLEN:alt長度 -ref長度。
1 | # annotate svtype and svlen to the cmrg sv.vcf |
處理後,再使用 bcftools 壓縮和建立索引。
1 | python process.py <(zcat HG002_GRCh38_CMRG_SV_v1.00.vcf.gz) | bcftools -o annotated_HG002_GRCh38_CMRG_SV_v1.00.vcf.gz -O z |
怎麼沒看到 CNVs 和 Tandem repeats 的效度評估?
CNVs 是結構變異的子類,評估方法於結構變異相同,只需從 truth & query vcf 篩選符合 CNVs 定義的變異。通常 CNVs 是指重複和缺失,不過像是 Dragen 團隊在評估 Dragen caller 時,則是把 CNVs 定義為長度 1000 bp 以上插入或刪除。要留意的是,因為 HG002 標準資料不含可靠的重複變異,目前只能用來評估刪除。另外,如果變異分析工具沒有提供明確的 alt 序列便無法比較插入序列相似度,使用 Truvari 時要記得把相關設置關掉 (--pctseq 0),以免低估效度。
1 | # 手邊沒電腦,用 AI 生成一段看起來的可以用的程式碼 |
而 Tandem repeats 則是指基因體中重複出現的鹼基組合(例如 ATT 重複三次成為 ATTATTATT),依照它重複單位的種類、次數、排列與長度可劃分為不同種類。評估效度時,通常會把不同種類的 tandem repeats 化約為插入或刪除,然後用結構變異的方式分析。
Dragen tandem repeats VCF 格式不符標準,所以評估效度前需要自行調整並計算 SVTYPE 和 SVLEN。
1 | chr1 1585949 . A <STR17>,<STR18> . PASS END=1585976;REF=9;RL=27;RU=AAT;VARID=chr1_1585949_1585976;REPID=chr1_1585949_1585976 GT:SO:REPCN:REPCI:ADSP:ADFL:ADIR:LC 1/2:SPANNING/SPANNING:17/18:17-17/18-18:11/6:16/16:0/0:39.689562 |
詳細作法可參考 DRAGEN 資料處理指南 truvari_giabtr_testing.pdf,簡言之:
- 轉換格式為 VCF v4.4,將
alt的格式從轉換為 CNV:TR - 保留
FILTER=PASS的變異 - 拆分 multi-allelic 變異
- 移除重複的變異
- 依照 tandem repeat 的重複單位和次數推論
alt的確切序列 - 添加
SVTYPE和SV資訊 - 對照參考基因體,取得
ref的確切序列
最後會得到一個與 Truvari 相容的格式,就能拿去跑分析了。
1 | chr1 1585949 chr1_1585949_1585976 AAATAATAATAATAATAATAATAATAAT AAATAATAATAATAATAATAATAATAATAATAATAATAATAATAATAATAAT SVTYPE=INS;SVLEN=24;RUS=AAT GT:CN 1/0:3.88889 |
結論
結構變異的偵測和評估技術還在快速發展,可以參考以下資源了解操作方法:
DRAGEN 團隊的研究論文除了使用標準資料評估自家工具的分析效度,還納入了多工具比較的情境,可作為評估自家算法或產品的指南。
- Behera et al. (2024). Comprehensive genome analysis and variant detection at scale using DRAGEN. Nature Biotechnology, 1-15.
- Behera. DRAGEN analysis. GitHub https://github.com/srbehera/DRAGEN_Analysis/ (2024).
標準資料的研究論文除了說明資料來源和篩選手段以外,也會介紹他們推薦的效度評估方式。
- Wagner et al. (2022). Curated variation benchmarks for challenging medically relevant autosomal genes. Nature biotechnology, 40(5), 672-680.
- Zook et al. (2020). A robust benchmark for detection of germline large deletions and insertions. Nature biotechnology, 38(11), 1347-1355.
如果想要了解評估工具的參數和算法,可以讀工具的官方文件和論文。
- English et al. (2022). Truvari: refined structural variant comparison preserves allelic diversity. Genome Biology, 23(1), 271.
- English. Project Adotto. Github https://github.com/acenglish/truvari/ (2024).
- English. Project Adotto. GitHub https://github.com/ACEnglish/adotto/ (2024).
- Wan & Ho. Wittyer. GitHub https://github.com/Illumina/witty.er (2023). (witty.er 是 illumina 的變異分析評估工具,沒有正式的論文發表)