ROSALIND|RNA Splicing (SPLC)
給定一條 DNA 序列與其 intron 序列(兩者儲存於同一個 FASTA 檔案),求此序列經過轉錄、剪切與轉譯後所產生的蛋白質序列。
After identifying the exons and introns of an RNA string, we only need to delete the introns and concatenate the exons to form a new string ready for translation.
Given: A DNA string s (of length at most 1 kbp) and a collection of substrings of s acting as introns. All strings are given in FASTA format.
Return: A protein string resulting from transcribing and translating the exons of s. (Note: Only one solution will exist for the dataset provided.)
(https://rosalind.info/problems/splc/)
知識背景
在轉錄時,參與這個過程的酵素子單元與輔助蛋白質(例如 transcription factors)會附著到位於目標基因上游的 promoter 序列,組成以 RNA 聚合酶(RNA polymerase)為中心的轉錄複合體 1 。在 RNA 聚合酶與其它酵素的催化下,轉錄複合體會解開 DNA 的雙股螺旋並且打破其間的氫鍵,再以其中一股為模板(模板股,template strand),從 5’ 端至 3’ 端逐一添加與模板股互補的核苷酸來合成 mRNA,這個過程會持續到複合體抵達 terminator 序列為止。
轉錄的原始產物被稱為 precursor mRNA,它的序列組成與模板股的互補股(密碼股,coding strand)一致,除了其中的 Thymine 被 Uracil 所取代。在真核生物中,這些 precursor mRNA 會停留在細胞核內被剪切體(splicesome)進一步加工2,移除其中部分的序列,再把剩餘的片段連結成 mature mRNA,送往細胞質完成後續的轉譯。
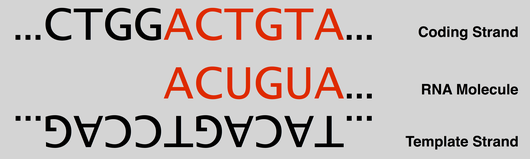
(圖片來源:https://rosalind.info/problems/splc/)
在剪切過程中,從 pre-mRNA 移除的片段被稱為 introns,而那些保留在 mature mRNA 的片段則叫做 exons。由於存在轉錄後加工,所以 DNA 序列無法透過轉譯密碼表直譯為蛋白質序列。此外,即使是 DNA 上相同的區域,在轉錄後也可能因為剪接的方式不同(alternative splicing),而產生相異的 mature mRNA,使得同一段 DNA 序列可以對應到多種蛋白質。
依據 DNA 片段在轉錄與轉譯後加工的狀況,真核生物的基因體可以劃分出以下幾種結構:
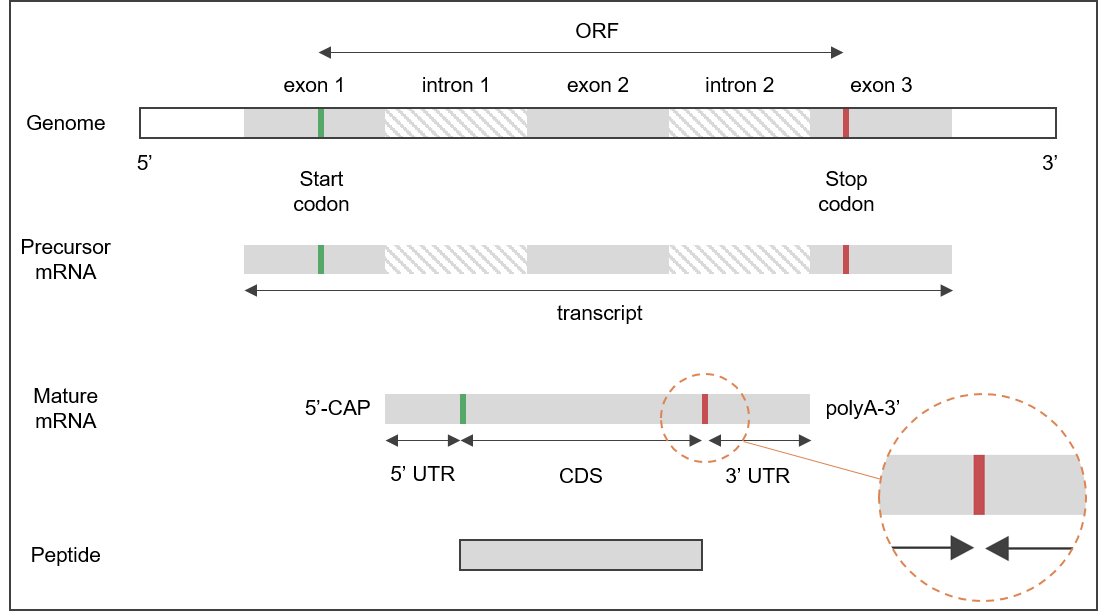
- ORF:起始密碼子(start codon)到終止密碼子(stop codon)之間的 DNA 片段,它是理論上能夠轉譯出蛋白質的區域。3
- intron:轉錄後加工時,從 precursor mRNA 移除的片段。
- exon:轉錄後加工後,保留在 mature mRNA 的片段。
- transcript:轉錄時合成的任何 mRNA 序列,包含 precursor mRNA 與 mature mRNA。由於同一段 DNA 可以對應多種 mature mRNA,所以 ensembl 等資料庫也會用 transcript 指稱 DNA 的特定剪接方式,以區分屬於不同 mature mRNA 的基因體註解資訊。
- CDS:coding sequence,mature mRNA 中實際轉譯出蛋白質的片段,它通常介於起始密碼子到終止密碼子(但不包含終止密碼子)之間。
- UTR:untranslated region, mature mRNA 5’ 和 3’ 兩端未經轉譯的片段。
我把這些片段的長度關係歸納如下:
- transcript = introns + exons
- exon = CDSs + 5’/3’ UTR + stop codon
- intron = transcript - exons
- CDSs = exons - 5’/3’ UTR - stop codon
這一題要求我們模擬轉錄、剪接與轉譯的過程,移除指定 DNA 當中的 introns,求出特定 transcript 對應的蛋白質序列。雖然題目裡沒有明確註記,但是測試序列必定是一個長度為三的倍數的 ORF,而且移除 introns 不會產生其它 ORFs。因此,作答時不需要考慮不同 readframe 的影響。
另外,雖然測試資料使用 FASTA 檔案儲存 DNA 和 introns 序列,可是它其實不適合儲存基因體結構資訊。首先,FASTA 檔案依賴自身以外的補充資訊來說明各序列的屬性。以這題為例,我們無法從測試資料得知何者為 DNA 或 intron,還得參考題目的說明。其次,只憑序列資訊難以區分位於目標基因內/外的 introns,這些重複的 intron 可能導致程式誤判。最後,序列比對不只較費時,紀錄完整序列也需要更多儲存空間,兩者都會影響程式執行效率。
因此,實務上會以 genome feature format(GTF/GRF) 儲存基因體各區域的屬性與特徵。GTF/GRF 是以 tab 分隔的文字檔案,包含 transcript 的名稱、特徵的類型、還有它在基因體的座標等欄位。因此,在比對樣本序列與參考基因體之後,即可透過 NCBI 的基因特徵資料庫配合座標資訊,標示出各序列的屬性。
解題觀念
這個練習的解題步驟依序為讀取 FASTA、移除 introns和轉譯 DNA。因為我在之前的文章已經解釋過部分功能,所以這裡只說明移除 intron 的方式,其它步驟的程式碼可參考我的 Github。
剪接 DNA/mRNA 的問題,可以視為移除字串內多個子字串的問題4。我們可以用多次字串取代(str.replace())或是用一次正則表達式,把 intron 字串抽換為空字串。
這兩種策略的運算效率相去不遠5,不過如果採用多次字串取代的策略,需要考量執行順序與重複取代的問題。畢竟,DNA 序列會隨字串取代而變化,如果途中產生新的 intron 片段,就會造成額外的剪切。
Python 實作
以 str.replace() 實作
即遍歷所有 intron,以空字串取代 DNA 內相符的片段。
1 | def rm_introns(dna, introns): |
以正則表達式實作
Python 的正則表達式套件(re)可以處理多字串的比對、取代與分割。此處應用的是 substitution 功能,它的語法為 re.sub(r"<pattern>", new, str),其中 <pattern> 是擷取特定字串的正則表達式語法。以這題為例,我們可以透過 pattern_1|pattern_2|pattern_3 ( | 表示「或」) 來擷取任一指定字串。因為我預設 intron 以 list 輸入,所以才用 str.join() 建立字串擷取的正則表達式6。
因為許多生物資訊工具運行於 linux 命令列,所以我們常用正則表達式配合 grep、sed、awk 等工具,快速從 FASTA、BED、VCF 等容量龐大的檔案擷取所需的資訊,以檢視分析結果是否符合預期。儘管現在很多 AI 工具都能幫忙生成正則表達式,不過我認為如果要把命令寫成腳本,還是有必要學習表達式的規則,以了解程式碼運作的邏輯,避免軟體出現非預期的錯誤。
在搜尋引擎查詢 regex introduction 即可找到許多學習資源,其中我特別喜歡 Regular Expressions for Regular Folk 的編排。如果自認已經掌握正則表達式的精隨,可以移步 Regex Golf 挑戰用最短的式子擷取題目指定的匹配字串。
1 | import re |
- 1.轉譯啟動機制的詳細說明可參考 Clancy. (2008) DNA transcription. Nature Education. 的 Transcription Initiation 一節。↩
- 2.RNA 裁切機制的詳細說明可參考 Clancy. (2008) RNA Splicing: Introns, Exons and Spliceosome. Nature Educaiton.。↩
- 3.這張示意圖只呈現了其中一種 mRNA 的剪接方式。在其他剪接方式下,終止密碼子可能落在範例 ORF 的內部,對應著長度較範例短的蛋白質。↩
- 4.可以參考 Replacing Multiple Patterns in a Single Pass 或 How to replace multiple substrings of a string? 等討論。↩
- 5.依據 Fastest implementation to do multiple string substitutions in python 的測試,使用正則表達式或多重 str.replace() 的表現到長度數百萬的字串仍難分軒輊。因此從效能角度來看,這兩種方式對於幾 kb 的 DNA 字串其實沒有太大的差異。↩
- 6.這題也可以用 re.split(),以 introns 為分隔符切斷 DNA 序列,再用 str.joint() 合併剩餘的序列。↩