最佳化 DADA2 的定序資料品質控管參數
在微生物增幅子分析當中,DADA2 是常用於校正 illumina 定序錯誤的軟體。在使用這軟體前,需要移除和裁切低品質的定序產物,確保資料在可校正的範圍之內。對於讀長 300 bp 的 illumina 雙端定序產物而言,可以選擇的參數非常多。目前參數的選擇往往依據經驗或是直接套用預設值,但是所以我想設計一種,能依據資料特性來選擇最佳參數的方法,改善分析過程的解釋性和把握度。
我設想的方法是 FIGARO (Weinstein et al. 2019.) 的延伸,大方向是先依照定序產物和校正演算法的特性,限縮可能的選項,再依照研究需求來排序不同參數的優劣。
品質控管的目標是盡可能保障定序產物的品質,又能保留足夠讀數的序列量,這在講究數據代表性的微生物生態研究相當重要。由於 illumina 定序的品質會隨長度遞減,而且序列的品質分數會在 DADA2 校正後遺失,所以冗長的序列對後續資料處理沒有幫助。
因此,可以把裁切方式限縮到足以保證兩股序列能順利合併的最小長度即可。關於合併雙端定序產物所需的最小長度,可參考 Edgar 的見解:Long overlaps are not needed, so 2x250 can do better than V4)。另一方面,雙端定序產物也要保持長度一致才能輸入 DADA2 演算法,以免推論 ASVs 的標準不一致。因此,裁切序列的方式只有有限的組合,其關係為:裁切方式 = 增幅區域長 + 重疊區域長 – 序列讀長。

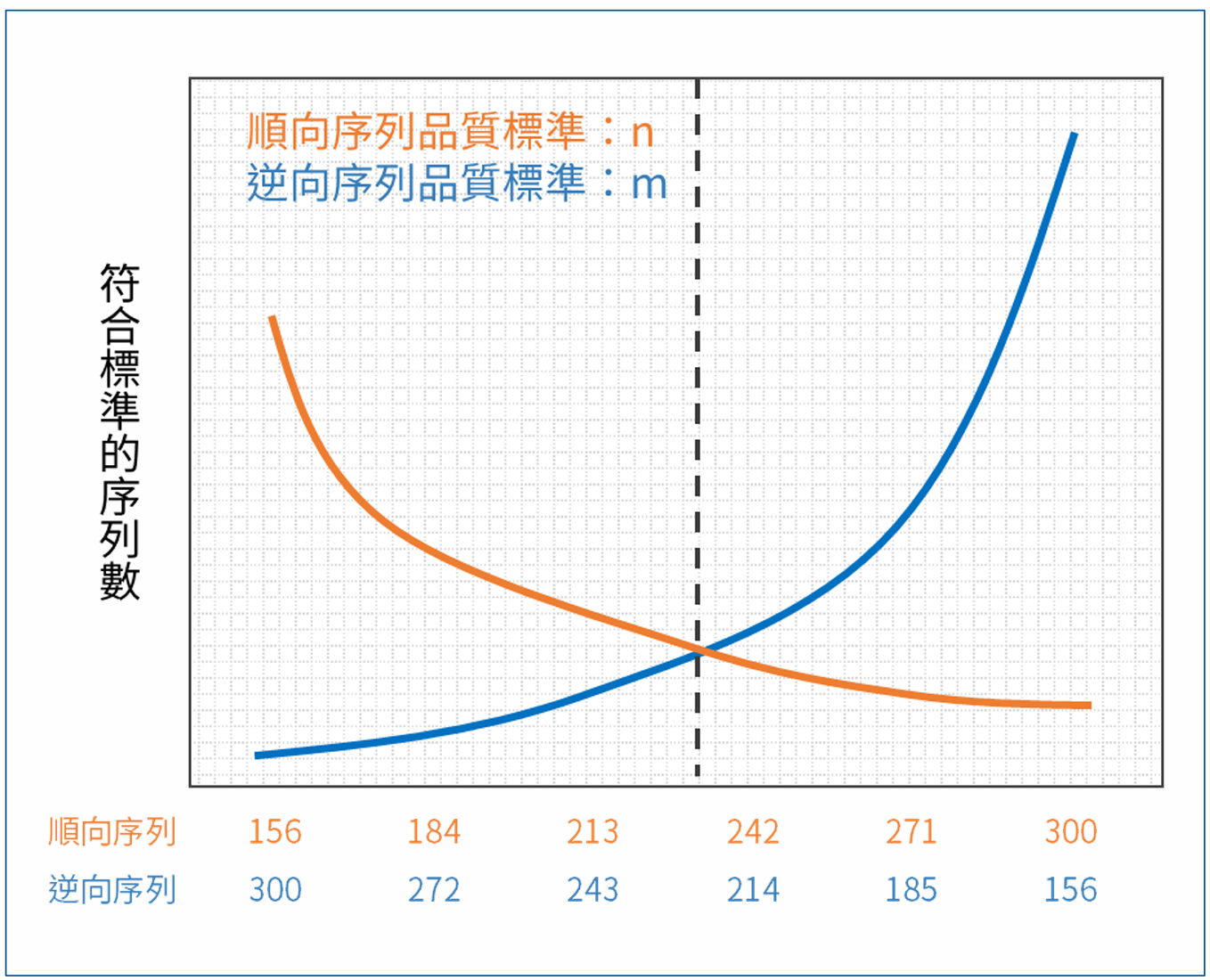
因為僅存的一股無法在後續步驟順利合併,所以通過篩選的序列數取決於兩股序列中品質較差的那一股。對於任意品質篩選標準,我們都能預期存在一組能保留最多序列的裁切方式。
下圖的橫軸是兩股序列裁切後的長度,縱軸則是通過特定品質篩選標準的序列數。通過標準的序列數隨長度遞減,兩條關係線的交叉處即是在該標準下最佳的長度裁切方式。
最後,將不同參數組合的通過率和篩選標準作圖,便能得出兩者的關係。可以發現,序列通過率的增幅會隨標準放寬遞減。我認為,合適的參數能在確保定序品質的情況下,保留更多的序列。所以,可以選擇序列通過率增幅的極值,作為選擇參數的依據。
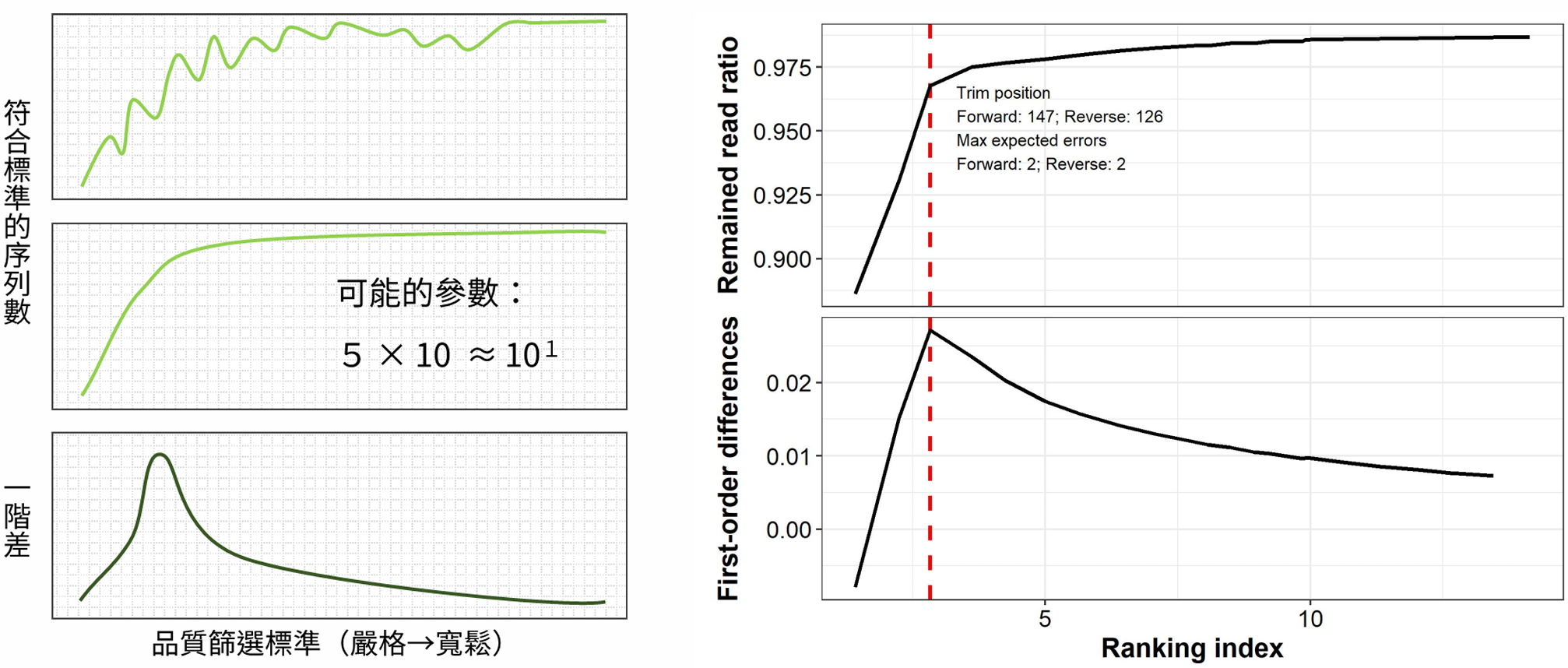
以圖右的示範資料為例,這表示在這方法推薦的參數組合下,能確保序列品質錯誤率在兩個 bp 以下,又能保留九成五以上的序列。
執行此算法的程式碼可參考我的 Github optimize_dada2_qc。當初因為急用,所以沒有改善這支腳本的命令列使用體驗。另外,一開始我是用全部序列當作最佳化參數的資料,但這樣容易受到樣本深度不均的問題影響。因此,除了改善程式碼的使用體驗以外,也能新增抽樣功能,增加執行速度並且排除一些偏誤。又或是增設一些閾值,讓對於最低樣本深度有要求的用戶能夠決定自己願意犧牲多少品質來換取保留的序列數。
最後提及此方法跟 FIGARO 的設計差異。FIGARO 要求用戶指定篩選序列的標準,我的策略則是以保留的序列數來決定篩選和裁切方式。因此,用戶只要決定雙端定序產物的重疊區域長度,不用煩惱怎麼設定品質篩選的閾值。
Weinstein et al. (2019). FIGARO: An efficient and objective tool for optimizing microbiome rRNA gene trimming parameters. BioRxiv, 610394.